Хорея Гентингтона
Хорея Генгтингтона, или болезнь Генгтингтона - прогрессирущее генетическое заболевание нервной системы, которое вызывает неконтролируемые движения, эмоциональные проблемы и слабоумие.
Причины хореи Гентингтона
Болезнь Гентингтона вызывают мутации в гене HTT. Ген HTT содержит инструкции по созданию белка, называемого хантингтин.
Хотя функция этого белка неизвестна, он, по-видимому, играет важную роль в нервных клетках мозга - нейронах.
Мутация HTT, которая вызывает болезнь Гентингтона, включает сегмент ДНК, известный как тринуклеотидный повтор CAG. Этот сегмент состоит из серии трех строительных блоков ДНК (C-цитозин, A-аденин и G-гуанин), которые появляются несколько раз подряд. Обычно сегмент CAG повторяется от 10 до 35 раз в пределах гена. У людей с болезнью Гентингтона сегмент CAG повторяется от 36 до более 120 раз. Люди с 36-39 повторениями CAG могут иметь или не иметь признаки и симптомы болезни Гентингтона, в то время как у людей с 40 или более повторениями почти всегда развивается расстройство.
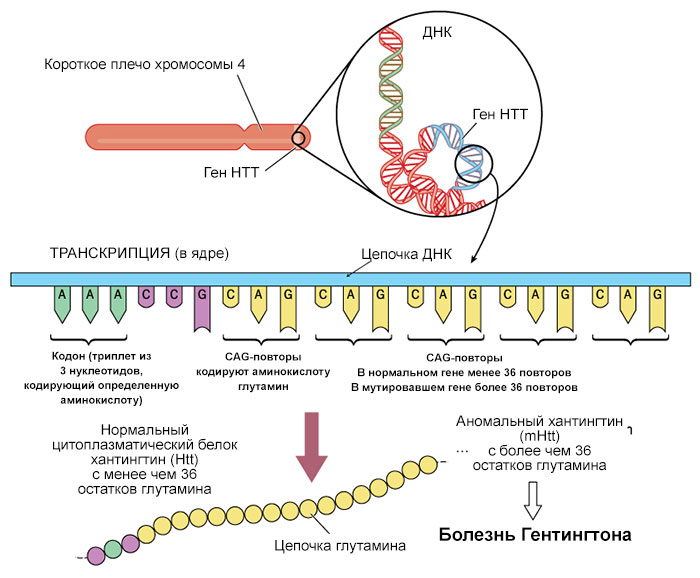
Увеличение размера сегмента CAG приводит к образованию аномально длинной версии белка хантингтина. Удлиненный белок разрезается на более мелкие токсичные фрагменты, которые связываются вместе и накапливаются в нейронах, нарушая нормальные функции этих клеток. В основе cимптомов болезни Гентингтона, возможно, лежит гибель нейронов в определенных областях мозга.
Частота болезни Гентингтона
Болезнь Гентингтона поражает от 3 до 7 на 100 000 человек белой расы. Это состояние менее распространено среди некоторых других групп населения, включая людей японского, китайского и африканского происхождения.
Начало и симптомы хореи Гентингтона
Болезнь Гентингтона у взрослых, наиболее распространенная форма этого расстройства, обычно проявляется в возрасте 30-40 лет.
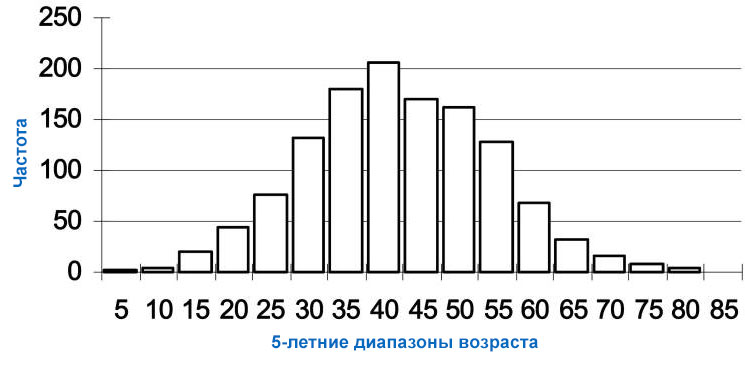
Ранние симптомы могут включать раздражительность, депрессию, небольшие непроизвольные движения, плохую координацию и проблемы с изучением новой информации или принятием решений. У многих людей с болезнью Гентингтона развиваются непроизвольные подергивания или подергивания, известные как хорея. По мере прогрессирования заболевания эти движения становятся более выраженными. Люди с этим состоянием люди могут иметь проблемы с ходьбой, речью и глотанием, они также подвержены психические расстройства, снижение умственных способностей. Люди с начальной формой болезни Гентингтона обычно живут примерно через 15-20 лет после появления признаков и симптомов.
Менее распространенная форма болезни Гентингтона, известная как ювенильная форма, начинается в детстве или в подростковом возрасте. также включает проблемы с движением и психические и эмоциональные изменения. Дополнительные признаки ювенильной формы включают медленные движения, неуклюжесть, частое падение, ригидность, невнятная речь и слюнотечение. Школьная успеваемость снижается, так как способность мыслить ослабевает. У 30-50% детей с этим заболеванием случаются судороги. Ювенильная болезнь Гентингтона имеет тенденцию прогрессировать быстрее, чем у взрослых, больные люди обычно живут 10-15 лет после появления признаков и симптомов.
Наследование хореи Гентингтона
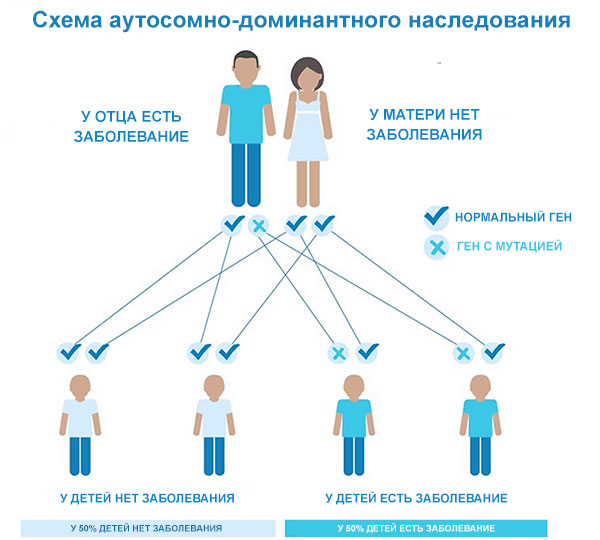
Это состояние наследуется по аутосомно-доминантному типу, что означает, что одной копии измененного гена в каждой клетке достаточно, чтобы вызвать расстройство. Больной хореей Гентингтона человек обычно наследует измененный ген от одного пострадавшего родителя. В редких случаях у человека с болезнью Гентингтона нет родителя с таким расстройством.
Риск для потомства больного - 50%.
Поскольку измененный ген HTT передается от одного поколения к другому, размер тринуклеотидного повтора CAG часто увеличивается в размерах. Большее количество повторов обычно связано с более ранним появлением симптомов. Это явление называется ожиданием. Люди с начальной взрослой формой болезни Гентингтона обычно имеют от 40 до 50 CAG-повторов в гене HTT, в то время как у людей с ювенильной формой расстройства, как правило, более 60 CAG-повторов.
Люди, у которых от 27 до 35 CAG-повторов в гене HTT, не заболевают болезнью Гентингтона, но у них есть риск иметь детей, у которых будет развиваться
расстройство. По мере того, как ген передается от родителя к ребенку, размер тринуклеотидного повтора CAG может удлиняться до диапазона, связанного с болезнью Гентингтона (36 повторов или более).
Гентингтоноподобное заболевание 2 типа (ГПЗ 2)
Гентингтоноподобное заболевание 2 типа (ГПЗ 2) – это наследственное прогрессирующее нейродегенеративное заболевание, клинически проявляющееся нарушениями в двигательной, когнитивной и психической сферах. Гентингтоноподобное заболевание 2 типа составляет около 1% случаев гентингтоноподобных заболеваний. Считается, что эта патология чаще встречается у лиц африканского происхождения.
Клинически гентингтоноподобное заболевание 2 типа не отличается от болезни Гентингтона, причиной которой является увеличение копий кодона СAG в гене HTT. При ГПЗ2 данная мутация отсутствует, однако отмечается экспансия CТG-повторов во 2А экзоне гена JPH3, кодирующего белок юнктофилин-3. В норме данный белок является компонентом комплексов, обеспечивающих связь ионных каналов и поверхности клетки.
Диагноз считается подтвержденным, если выявляется 40 и более повторов CTG в гене JPH3. Точный патогенез развития заболевания неизвестен, однако существует три различных гипотезы: полиаминокислотная токсичность, нарушение функции белка, нейротоксичность формируемой мРНК.
Как уже было сказано, клинически данное заболевание не отличается от болезни Гентингтона. К основным симптомам относятся:
*моторные нарушения: хорея, дистония, брадикинезия, тремор, ригидность;
*нарушение когнитивной функции: деменция (развивается через 10-15 лет от начала заболевания);
*психические нарушения: преимущественно депрессия, агрессивность.
*При анализе крови у части пациентов может быть выявлен акантоцитоз – появление в периферической
*крови эритроцитов со множественными выростами цитоплазмы.
При МРТ головного мозга отмечается атрофия базальных ганглиев (хвостатое ядро, скорлупа, бледный шар), уменьшение объема коры. По данным инструментальных исследований также не удается отличить болезнь Гентингтона от гентингтоноподобного заболевания 2 типа, однако отмечается, что лобная доля и substantia nigra чаще поражаются при последнем.
Тип наследования: аутосомно-доминантный. Для данного заболевания характерен феномен антиципации, то есть тяжесть клинических проявлений нарастает от поколения к поколению. Клиническая картина, а также возраст первого проявления заболевания коррелирует с размером экспансии.
Метод исследования:
- ПЦР-реакция экспансии CAG-повтора в гене HTT и экспансии CТG-повторов во 2А экзоне гена JPH3
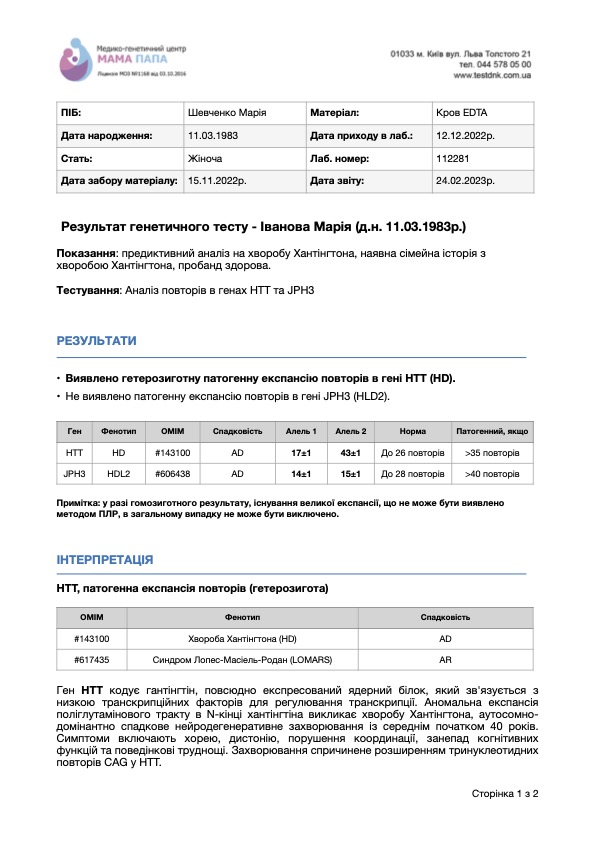
Образец заключения теста на Хорею Гентингтона

Как пройти исследование
Требования сдачи крови натощак нет (легкий завтрак и незадолго до взятия крови выпить 1-2 стакана обычной негазированной воды; детей обязательно поить негазированной водой порциями, до 150-200 мл, в течение 30 минут), в день сдачи крови не употреблять медикаменты (включая витамины и БАД), накануне исключить прием жареной и жирной пищи.
Проводится забор венозной крови в пробирку с ЭДТА.
Рекомендовано предоставить выписку/заключение врача, заполнить бланк заказа.
Во всех случаях по результатам необходимо получить консультацию врача-генетика.
 Киев
Киев
