Хорея Гентінгтона
Хорея Генгтингтона, або хвороба Генгтингтона - прогресуюче генетичне захворювання нервової системи, яке викликає неконтрольовані рухи, емоційні проблеми та недоумство.
Причини хореї Гентінгтона
Хвороба Гентінгтона викликають мутації в гені HTT. Ген HTT містить інструкції щодо створення білка, що називається хантінгтін.
Хоча функція цього білка невідома, він, мабуть, відіграє важливу роль у нервових клітинах мозку – нейронах.
Мутація HTT, що викликає хворобу Гентінгтона, включає сегмент ДНК, відомий як тринуклеотидний повтор CAG. Цей сегмент складається з серії трьох будівельних блоків ДНК (C-цитозин, A-аденін та G-гуанін), які з'являються кілька разів поспіль. Зазвичай сегмент CAG повторюється від 10 до 35 разів у межах гена. У людей із хворобою Гентінгтона сегмент CAG повторюється від 36 до більше 120 разів. Люди з 36-39 повтореннями CAG можуть мати або не мати ознак і симптомів хвороби Гентінгтона, у той час як у людей з 40 або більше повтореннями майже завжди розвивається розлад.
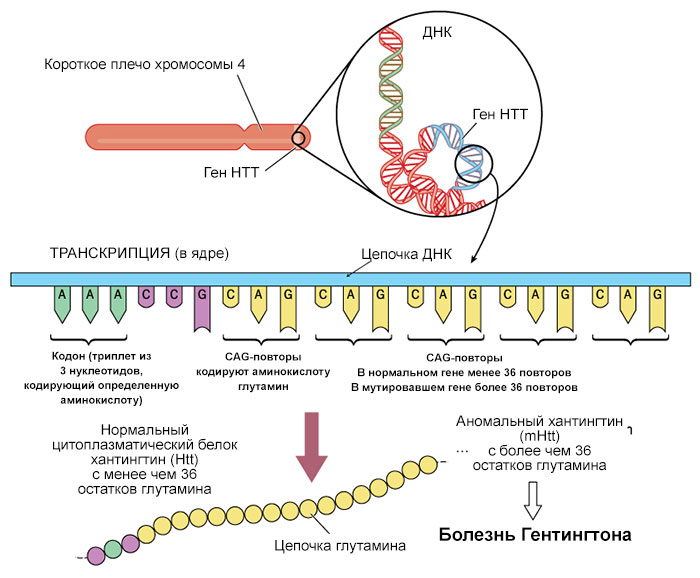
Збільшення розміру сегмента CAG призводить до утворення аномально довгої версії хантінгтіну білка. Подовжений білок розрізається на дрібніші токсичні фрагменти, які зв'язуються разом і накопичуються в нейронах, порушуючи нормальні функції цих клітин. В основі симптомів хвороби Гентінгтона, можливо, лежить загибель нейронів у певних областях мозку.
Частота хвороби Гентінгтона
Хвороба Гентінгтона вражає від 3 до 7 на 100 000 людей білої раси. Цей стан менш поширений серед інших груп населення, включаючи людей японського, китайського і африканського походження.
Початок і симптоми хореї Гентінгтона
Хвороба Гентінгтона у дорослих, найбільш поширена форма цього розладу, зазвичай проявляється у віці 30-40 років.
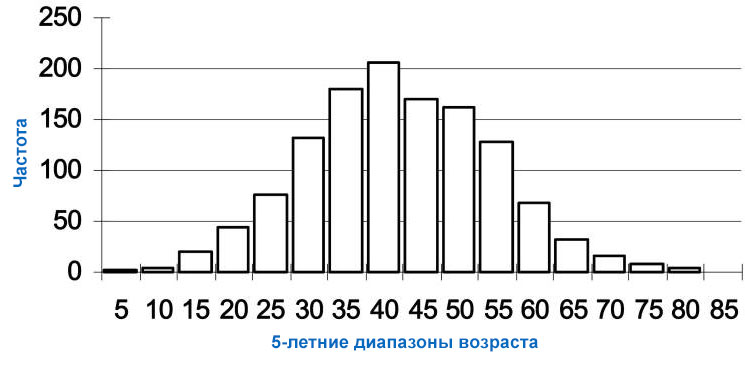
Ранні симптоми можуть включати дратівливість, депресію, невеликі мимовільні рухи, погану координацію та проблеми з вивчення нової інформації або прийняття рішень. У багатьох людей із хворобою Гентінгтона розвиваються мимовільні посмикування або посмикування, відомі як хорея. У міру прогресування захворювання ці рухи стають більш вираженими. Люди з цим станом люди можуть мати проблеми з ходьбою, промовою та ковтанням, вони також схильні до психічних розладів, зниження розумових здібностей. Люди з початковою формою хвороби Гентінгтона зазвичай живуть приблизно через 15-20 років після появи ознак та симптомів.
Менш поширена форма хвороби Гентінгтона, відома як ювенільна форма, починається в дитинстві або в підлітковому віці. також включає проблеми з рухом та психічні та емоційні зміни. Додаткові ознаки ювенільної форми включають повільні рухи, незграбність, часте падіння, ригідність, невиразне мовлення та слинотеча. Шкільна успішність знижується, оскільки здатність мислити слабшає. У 30-50% дітей із цим захворюванням трапляються судоми. Ювенільна хвороба Гентінгтон має тенденцію прогресувати швидше, ніж у дорослих, хворі люди зазвичай живуть 10-15 років після появи ознак і симптомів.
Спадкування хореї Гентінгтона
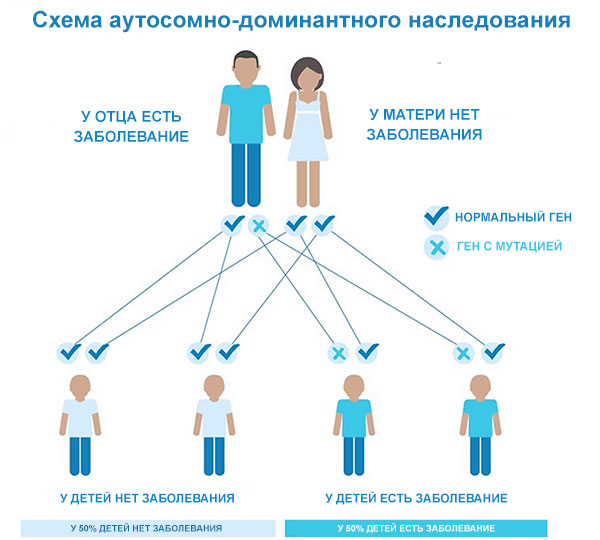
Цей стан успадковується за аутосомно-домінантним типом, що означає, що однієї копії зміненого гена в кожній клітині достатньо, щоб викликати розлад. Хвора на хорею Гентінгтона людина зазвичай успадковує змінений ген від одного постраждалого батька. У поодиноких випадках у людини з хворобою Гентінгтона немає батька з таким розладом.
Ризик для потомства хворого – 50%.
Оскільки змінений ген HTT передається від одного покоління до іншого, розмір тринуклеотидного повтору CAG часто збільшується у розмірах. Більша кількість повторів зазвичай пов'язані з більш раннім появою симптомів. Це називається очікуванням. Люди з початковою дорослою формою хвороби Гентінгтона зазвичай мають від 40 до 50 CAG-повторів у гені HTT, тоді як у людей з ювенільною формою розладу зазвичай більше 60 CAG-повторів.
Люди, у яких від 27 до 35 CAG-повторів у гені HTT, не хворіють на хворобу Гентінгтона, але у них є ризик мати дітей, у яких розвиватиметься
розлад. У міру того, як ген передається від батька до дитини, розмір тринуклеотидного повтору CAG може подовжуватися до діапазону, пов'язаного із хворобою Гентінгтона (36 повторів або більше).
Гентінгтоноподібне захворювання 2 типу (ГПЗ 2)
Гентінгтоноподібне захворювання 2 типу (ГПЗ 2) – це спадкове прогресуюче нейродегенеративне захворювання, що клінічно виявляється порушеннями у руховій, когнітивній та психічній сферах. Гентингтоноподібне захворювання 2 типу становить близько 1% випадків гентингтоноподібних захворювань. Вважається, що ця патологія найчастіше зустрічається в осіб африканського походження.
Клінічно гентингтоноподібне захворювання 2 типу не відрізняється від хвороби Гентингтона, причиною якої є збільшення копій кодону СAG у гені HTT. При ГПЗ2 ця мутація відсутня, але відзначається експансія CТG-повторів в 2А екзон гена JPH3, що кодує білок юнктофілін-3. У нормі цей білок є компонентом комплексів, що забезпечують зв'язок іонних каналів та поверхні клітини.
Діагноз вважається підтвердженим, якщо виявляється 40 і більше повторів CTG у гені JPH3. Точний патогенез розвитку захворювання невідомий, проте існує три різні гіпотези: поліамінокислотна токсичність, порушення функції білка, нейротоксичність мРНК, що формується.
Як уже було сказано, клінічно це захворювання не відрізняється від хвороби Гентінгтона. До основних симптомів відносяться:
*моторні порушення: хорея, дистонія, брадикінезія, тремор, ригідність;
* порушення когнітивної функції: деменція (розвивається через 10-15 років від початку захворювання);
*психічні порушення: переважно депресія, агресивність.
*При аналізі крові у частини пацієнтів може бути виявлено акантоцитоз – поява в периферичній
*крові еритроцитів з множинними виростами цитоплазми.
При МРТ головного мозку відзначається атрофія базальних гангліїв (хвостате ядро, шкаралупа, бліда куля), зменшення об'єму кори. За даними інструментальних досліджень також не вдається відрізнити хворобу Гентінгтона від гентингтоноподібного захворювання 2 типу, однак зазначається, що лобова частка і substantia nigra частіше уражаються при останньому.
Тип успадкування: аутосомно-домінантний. Для цього захворювання характерний феномен антиципації, тобто тяжкість клінічних проявів наростає від покоління до покоління. Клінічна картина, а також вік першого прояву захворювання корелює із розміром експансії.
Метод дослідження:
- ПЛР-реакція експансії CAG-повтору в гені HTT та експансії CТG-повторів у 2А екзоні гена JPH3
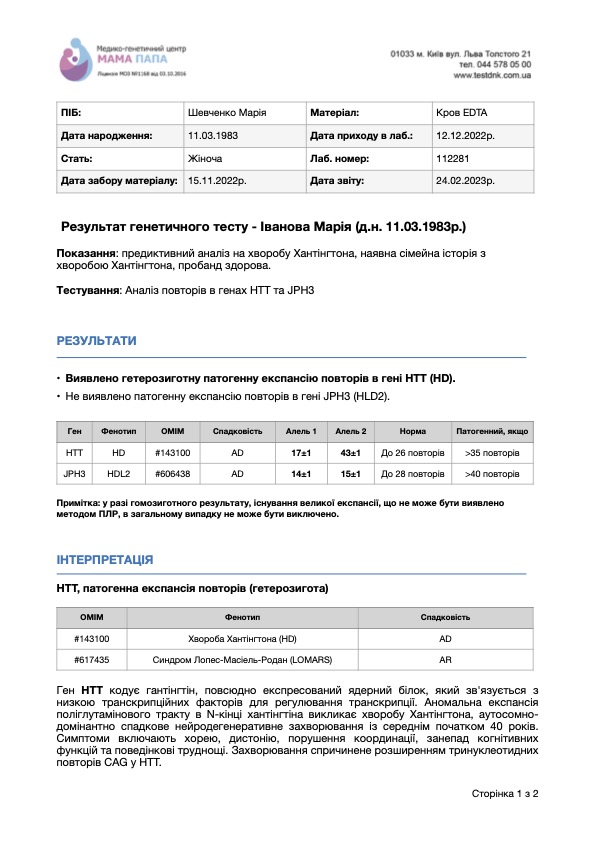
Образец заключения теста на Хорею Гентингтона

Як пройти дослідження
Вимоги здачі крові натще немає (легкий сніданок і незадовго до взяття крові випити 1-2 склянки звичайної негазованої води; дітей обов'язково напувати негазованою водою порціями, до 150-200 мл, протягом 30 хвилин), в день здачі крові не вживати медикаменти (включаючи вітаміни та БАД), напередодні виключити прийом смаженої та жирної їжі.
Проводиться забір венозної крові в пробірку з ЕДТА.
Рекомендовано надати виписку/висновок лікаря, заповнити бланк замовлення.
У всіх випадках за результатами необхідно отримати консультацію лікаря-генетика.

 Київ
Київ
