Панель из 37 генов мтДНК с чрезвычайно высоким охватом секвенирования.
Идеально подходит для пациентов с подозрением на митохондриальное заболевание, у которых был получен отрицательный результат с использованием целевой панели ядерных генов или тестирования всего экзома, который не включал митохондриальную ДНК.
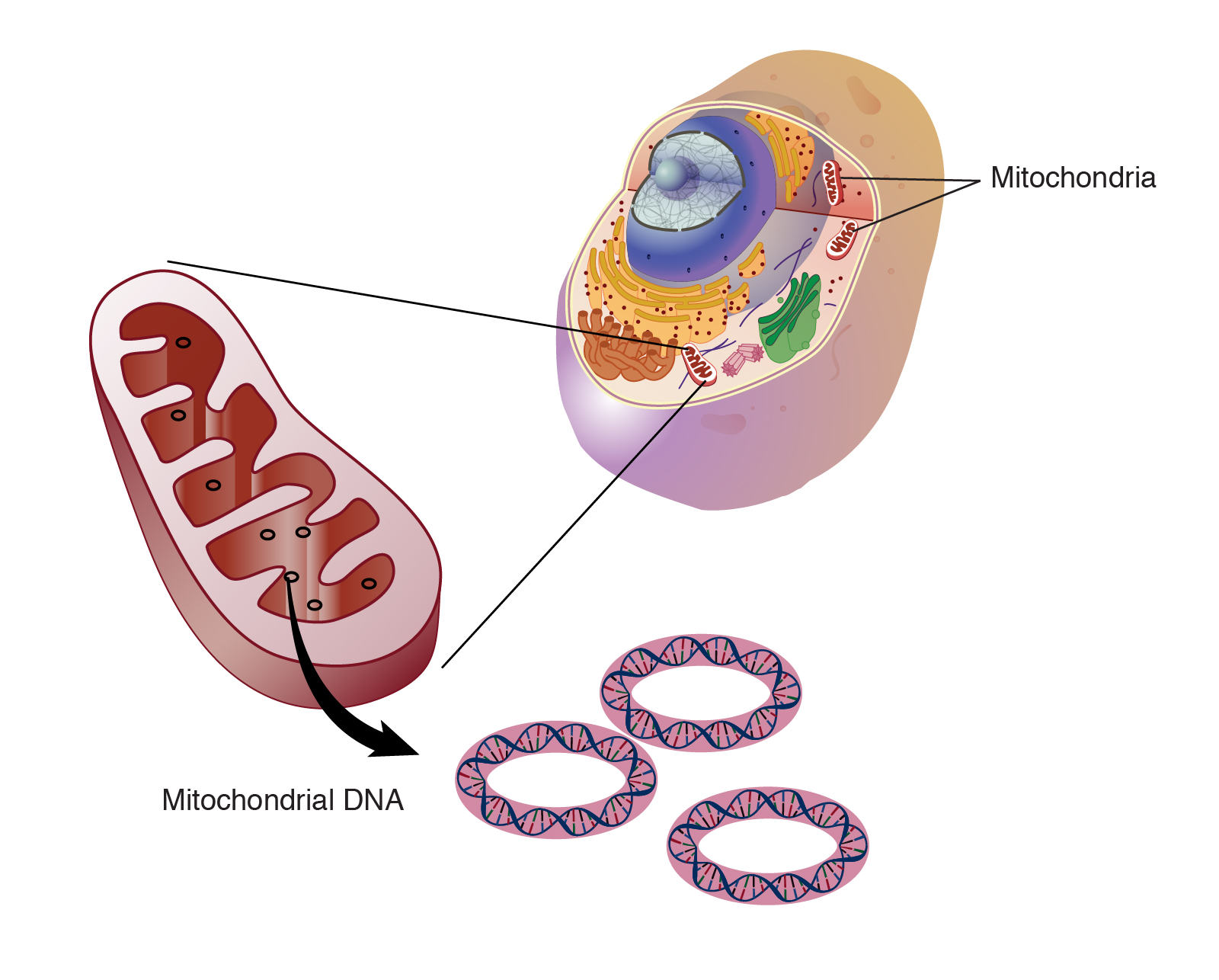
Эта панель включает митохондриальный геном (мтДНК) размером 16,5 т.п.н., содержащий 37 генов, все из которых необходимы для нормальной функции митохондрий. Тринадцать из этих генов кодируют полипептиды, образующие структурные субъединицы дыхательной цепи (комплексы RC I, III, IV и V), что является функционально важным и эволюционно ограниченным, а также РНК, необходимую для трансляции мтДНК, а именно 2 рРНК (MT- RNR1 и MT-RNR2, кодирующие 12S и 16S рРНК) и 22 транспортные РНК (тРНК, например, tRNALys), которые расположены между генами, кодирующими белок. Подавляющее большинство из более чем 1000 митохондриальных белков кодируются ядерными генами.
Есть несколько уникальных свойств, связанных с митохондриальным геномом, которые важны для понимания первичного заболевания митохондриальной ДНК:
1) в каждой клетке имеется несколько копий мтДНК;
2) мтДНК наследуется по материнской линии,
3) мутантная мтДНК сосуществует с мтДНК дикого типа (гетероплазмия),
4) необходима минимальная критическая доля мутантных мтДНК, прежде чем станет очевидной тканевая дисфункция (пороговый эффект),
5) мутационная нагрузка (уровень гетероплазмии) может варьировать среди различных тканей.
Митохондриальные заболевания представляют собой клинически гетерогенную группу заболеваний, возникающих в результате дисфункции митохондриальной дыхательной цепи. В то время как некоторые митохондриальные расстройства затрагивают только один орган, многие затрагивают несколько систем органов. Симптомы у пациентов могут варьироваться от легких до тяжелых и могут возникать в любом возрасте. Общие клинические признаки митохондриального заболевания включают утомляемость, слабость, метаболические инсульты, судороги, кардиомиопатию, аритмии, нарушения развития или когнитивные функции, сахарный диабет, нарушения слуха, зрения, роста, функции печени, желудочно-кишечного тракта или почек и многое другое.
Типичные митохондриальные заболевания с ранним началом включают синдром Ли, синдром истощения, синдром Кернса-Сейра (KSS) и синдром Пирсона, тогда как хроническая прогрессирующая наружная офтальмоплегия (CPEO), наследственная оптическая нейропатия Лебера (LHON), невропатия, атаксия и пигментный ретинит (NARP), митохондриальная энцефалопатия, лактоацидоз и инсультоподобные эпизоды (MELAS) наблюдаются в более позднем детстве или во взрослой жизни. На сегодняшний день зарегистрировано около 400 мутаций, которые, как известно, вызывают спектр митохондриальных заболеваний. Большинство изменений мтДНК представляют собой нейтральные полиморфизмы, которые определяют различные гаплогруппы населения и использовались, например, для отслеживания миграции людей.
Наследование мутаций в митохондриальном геноме носит особый характер. Если гены, заключенные в ядерной ДНК, дети получают поровну от обоих родителей, то митохондриальные гены передаются потомкам только от матери. Это связано с тем, что всю цитоплазму с содержащимися в ней митохондриями потомки получают вместе с яйцеклеткой, в то время как в сперматозоидах цитоплазма практически отсутствует. По этой причине женщина с митохондриальным заболеванием передаёт его всем своим детям, а больной мужчина - нет.
В нормальных условиях все митохондрии в клетке имеют одинаковую копию ДНК - гомоплазмия. Однако в митохондриальном геноме могут происходить мутации, и вследствие параллельного существования мутированной и немутированной мтДНК возникает гетероплазмия.
К настоящему времени известно более 200 заболеваний, вызванных мутацией мтДНК.
Клинические проявления митохондриальной патологии:
В случаях, когда человек с мутацией в митохондриальном гене несет смесь нормальной и мутантной ДНК - мутации поначалу могут вообще не иметь внешних проявлений. Нормальные митохондрии до поры до времени обеспечивают клетки энергией, компенсируя недостаточность функции митохондрий с дефектами. На практике это проявляется более или менее длительным бессимптомным периодом при многих митохондриальных заболеваниях. Однако рано или поздно наступает момент, когда дефектные формы накапливаются в количестве, достаточном для проявления патологических признаков. Возраст манифестации заболевания варьирует у разных больных. Раннее начало заболевания приводит к более тяжелому течению и неутешительному прогнозу.
Митохондриальные мутации проявляются широким рядом клинических симптомов. Эти мутации способны вовлекать тРНК, рРНК или структурные гены и могут выражаться биохимически как дефекты всей электронно-транспортной цепи или как дефекты отдельных энзимов. Митохондриальные цитопатии поражают множественные органные системы, но, как указывалось, предпочтительно поражаются органы с высокой метаболической активностью - мозг и скелетные мышцы. Таким образом, скелетные мышцы являются тканью выбора для выявления митохондриальных болезней.
Характерные признаки митохондриальных цитопатий:
- скелетные мышцы: низкая толерантность к физической нагрузке, гипотония, проксимальная миопатия, включающая фациальные и фарингеальные мышцы, офтальмопарез, птоз;
- сердце: нарушения сердечного ритма, гипертрофическая миокардиопатия;
- центральная нервная система: атрофия зрительного нерва, пигментная ретинопатия, миоклонус, деменция, инсультоподобные эпизоды, расстройства психики;
- периферическая нервная система: аксональная нейропатия, нарушения двигательной функции гастроинтестинального тракта;
- эндокринная система: диабет, гипопаратиреоидизм, нарушение экзокринной функции панкреас, низкий рост.
Гены, входящие в панель:
| Ген |
Ассоциированный фенотип |
Наслед.* |
ClinVar** |
HGMD** |
|
MT-ATP6
|
Нейропатия, атаксия и пигментный ретинит, наследственная оптическая невропатия Лебера, атаксия и полинейропатия, с началом во взрослом возрасте, кардиомиопатия, инфантильная гипертрофическая, синдром Ли, стриатонигральная дегенерация, инфантильная, митохондриальная |
Mitochondrial |
19 |
|
|
MT-ATP8
|
Кардиомиопатия, апикальная гипертрофическая и нейропатия, Кардиомиопатия, инфантильная гипертрофическая | Mitochondrial |
4 |
|
|
MT-CO1
|
Миоглобинурия, рецидивирующая, наследственная оптическая нейропатия Лебера, сидеробластная анемия, дефицит цитохром-С-оксидазы, глухота, митохондриальная |
Mitochondrial |
17 |
|
|
MT-CO2
|
Дефицит цитохром C оксидазы |
Mitochondrial |
8 |
|
|
MT-CO3
|
Дефицит цитохром C оксидазы, наследственная оптическая нейропатия Лебера | Mitochondrial |
9 |
|
|
MT-CYB
|
|
Mitochondrial |
69 |
|
|
MT-ND1
|
Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды, наследственная оптическая нейропатия Лебера, атрофия и дистония зрительного нерва Лебера |
Mitochondrial |
21 |
|
|
MT-ND2
|
Наследственная оптическая нейропатия Лебера, дефицит митохондриального комплекса I |
Mitochondrial |
6 |
|
|
MT-ND3
|
Атрофия и дистония зрительного нерва Лебера, дефицит митохондриального комплекса I |
Mitochondrial |
7 |
|
|
MT-ND4
|
Наследственная оптическая нейропатия Лебера, атрофия и дистония зрительного нерва Лебера, дефицит митохондриального комплекса I |
Mitochondrial |
11 |
|
|
MT-ND4L
|
Наследственная оптическая нейропатия Лебера |
Mitochondrial |
2 |
|
|
MT-ND5
|
Миоклоническая эпилепсия с рваными красными волокнами, митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды, наследственная оптическая нейропатия Лебера, дефицит митохондриального комплекса I |
Mitochondrial |
19 |
|
|
MT-ND6
|
Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды, онкоцитома, наследственная оптическая нейропатия Лебера, атрофия и дистония зрительного нерва Лебера, дефицит митохондриального комплекса I |
Mitochondrial |
16 |
|
|
MT-RNR1
|
Глухота, митохондриальная |
Mitochondrial |
3 |
|
|
MT-RNR2
|
Токсичность/резистентность к хлорамфениколу |
Mitochondrial |
2 |
|
|
MT-TA
|
|
Mitochondrial |
4 |
|
|
MT-TC
|
Митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды | Mitochondrial |
3 |
|
|
MT-TD
|
|
Mitochondrial |
1 |
|
|
MT-TE
|
Диабет-синдром глухоты, Митохондриальная миопатия, инфантильная, транзиторная, Митохондриальная миопатия при сахарном диабете |
Mitochondrial |
5 |
|
|
MT-TF
|
Миоклоническая эпилепсия с рваными красными волокнами, нефропатия, тубулоинтерстициальная, энцефалопатия, митохондриальная, эпилепсия, митохондриальная, миопатия, митохондриальная, митохондриальная энцефаломиопатия с лактоацидозом и инсультоподобными эпизодами |
Mitochondrial |
7 |
|
|
MT-TG
|
|
Mitochondrial |
3 |
|
|
MT-TH
|
|
Mitochondrial |
4 |
|
|
MT-TI
|
|
Mitochondrial |
7 |
|
|
MT-TK
|
Миоклоническая эпилепсия с рваными красными волокнами, синдром Лея |
Mitochondrial |
5 |
|
|
MT-TL1
|
Дефицит цитохром-с-оксидазы, миоклоническая эпилепсия с рваными красными волокнами, митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды, диабет-синдром глухоты, синдром циклической рвоты, СВДС |
Mitochondrial |
14 |
|
|
MT-TL2
|
Митохондриальное мультисистемное заболевание, прогрессирующая наружная офтальмоплегия, митохондриальная миопатия, митохондриальная энцефаломиопатия с лактоацидозом и инсультоподобными эпизодами |
Mitochondrial |
5 |
|
|
MT-TM
|
Синдром Лея, митохондриальное мультисистемное расстройство |
Mitochondrial |
1 |
|
|
MT-TN
|
Прогрессирующая наружная офтальмоплегия, митохондриальное мультисистемное заболевание |
Mitochondrial |
3 |
|
|
MT-TP
|
|
Mitochondrial |
2 |
|
|
MT-TQ
|
Митохондриальное мультисистемное расстройство |
Mitochondrial |
2 |
|
|
MT-TR
|
Энцефалопатия, митохондриальная |
Mitochondrial |
2 |
|
|
MT-TS1
|
Миоклоническая эпилепсия с рваными красными волокнами, митохондриальная миопатия, энцефалопатия, лактоацидоз и инсультоподобные эпизоды |
Mitochondrial |
10 |
|
|
MT-TS2
|
Митохондриальное мультисистемное расстройство |
Mitochondrial |
2 |
|
|
MT-TT
|
|
Mitochondrial |
5 |
|
|
MT-TV
|
Гипертрофическая кардиомиопатия (ГКМП), синдром Ли, митохондриальное мультисистемное заболевание, митохондриальная энцефаломиопатия с лактоацидозом и инсультоподобными эпизодами |
Mitochondrial |
3 |
|
|
MT-TW
|
Синдром Лея, миопатия, митохондриальная |
Mitochondrial |
8 |
|
|
MT-TY
|
Митохондриальное мультисистемное расстройство |
Mitochondrial |
4 |
|
* Тип наследования: аутосомно-доминантный (AD), аутосомно-рецессивный (AR), митохондриальный (mi), Х-сцепленный (XL), Х-сцепленный доминантный (XLD) и Х-сцепленный рецессивный (XLR);
** ClinVar, HGMD - количество вариантов гена, классифицированных как патогенные или вероятно патогенные в базе данных ClinVar, HGMD.
Преимущества этого теста
- Лаборатория (США), аккредитованная САР
- Персонал, сертифицированный CLIA, проводит клинические испытания в лаборатории, сертифицированной CLIA.
- Мощные технологии секвенирования, передовые методы обогащения мишеней и конвейеры точной биоинформатики обеспечивают превосходную аналитическую производительность.
- Тщательное составление клинически эффективных и научно обоснованных генных панелей
- Общедоступная аналитическая проверка, демонстрирующая полную информацию о производительности теста
- Анализ некодирующих вариантов, вызывающих заболевания
- Строгая схема классификации вариантов
- Систематический рабочий процесс клинической интерпретации с использованием проприетарного программного обеспечения, обеспечивающего точную и прослеживаемую обработку данных NGS.
- Подробнейшее клиническое заключение
Как пройти исследование
Требования сдачи крови натощак нет (легкий завтрак и незадолго до взятия крови выпить 1-2 стакана обычной негазированной воды; детей обязательно поить негазированной водой порциями, до 150-200 мл, в течение 30 минут), в день сдачи крови не употреблять медикаменты (включая витамины и БАД), накануне исключить прием жареной и жирной пищи, исключить за 32 часа алкоголь.
Проводится забор венозной крови в пробирку с ЭДТА 4 мл.
Необходимо предоставить (взять с собой или прислать нам на электронную почту) заключение врача с подробной клинической картиной (описание симтомов, диагноз/предварительный диагноз) и фото исследуемого пациента (если есть фенотипически значимые особенности), заполнить в электронном виде анкету.
Во всех случаях по результатам секвенирования необходимо получить консультацию врача-генетика.
 Киев
Киев
