Панель із 37 генів мтДНК із надзвичайно високим охопленням секвенування.
Ідеально підходить для пацієнтів з підозрою на мітохондріальне захворювання, які мали негативний результат з використанням цільової панелі ядерних генів або тестування всього екзома, який не включав мітохондріальну ДНК.
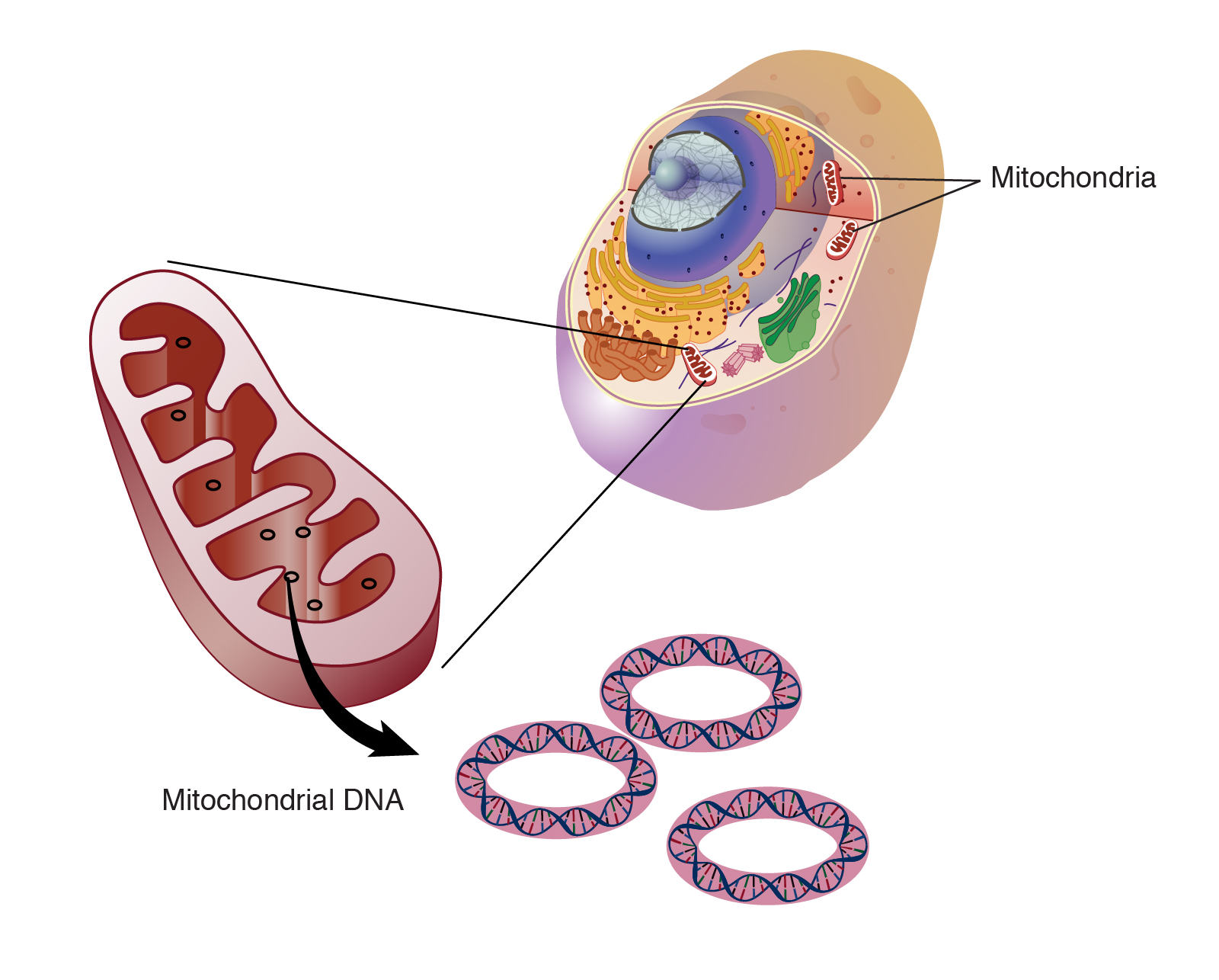
Ця панель включає мітохондріальний геном (мтДНК) розміром 16,5 т.п.н., що містить 37 генів, всі з яких необхідні для нормальної функції мітохондрій. Тринадцять з цих генів кодують поліпептиди, що утворюють структурні субодиниці дихального ланцюга (комплекси RC I, III, IV і V), що є функціонально важливим та еволюційно обмеженим, а також РНК, необхідну для трансляції мтДНК, а саме 2 рРНК (MT-RNR1 та MT-RNR2, що кодують 12S та 16S рРНК) та 22 транспортні РНК (тРНК, наприклад, tRNALys), які розташовані між генами, що кодують білок. Переважна більшість більш ніж 1000 мітохондріальних білків кодуються ядерними генами.
Є кілька унікальних властивостей, пов'язаних з мітохондріальним геномом, які важливі для розуміння первинного захворювання на мітохондріальну ДНК:
1) у кожній клітині є кілька копій мтДНК;
2) мтДНК успадковується по материнській лінії,
3) мутантна мтднк співіснує з мтднк дикого типу (гетероплазмія),
4) необхідна мінімальна критична частка мутантних мтДНК, перш ніж стане очевидною тканинна дисфункція (пороговий ефект),
5) мутаційне навантаження (рівень гетероплазмії) може варіювати серед різних тканин.
Мітохондріальні захворювання є клінічно гетерогенною групою захворювань, що виникають в результаті дисфункції мітохондріального дихального ланцюга. У той час як деякі мітохондріальні розлади торкаються лише одного органу, багато хто торкається кількох систем органів. Симптоми у пацієнтів можуть змінюватись від легень до тяжких і можуть виникати в будь-якому віці. Загальні клінічні ознаки мітохондріального захворювання включають стомлюваність, слабкість, метаболічні інсульти, судоми, кардіоміопатію, аритмії, порушення розвитку або когнітивні функції, цукровий діабет, порушення слуху, зору, росту, функції печінки, шлунково-кишкового тракту або нирок та багато іншого.
Типові мітохондріальні захворювання з раннім початком включають синдром Лі, синдром виснаження, синдром Кернса-Сейра (KSS) і синдром Пірсона, тоді як хронічна прогресуюча зовнішня офтальмоплегія (CPEO), спадкова оптична нейропатія Лебера (LHON), невропатия, ретиніт (NARP), мітохондріальна енцефалопатія, лактоацидоз та інсультоподібні епізоди (MELAS) спостерігаються у пізнішому дитинстві або у дорослому житті. На сьогоднішній день зареєстровано близько 400 мутацій, які, як відомо, спричиняють спектр мітохондріальних захворювань. Більшість змін мтДНК є нейтральними поліморфізмами, які визначають різні гаплогрупи населення і використовувалися, наприклад, для відстеження міграції людей.
Спадкування мутацій у мітохондріальному геномі має особливий характер. Якщо гени, ув'язнені в ядерної ДНК, діти одержують порівну від обох батьків, то мітохондріальні гени передаються нащадкам тільки матері. Це пов'язано з тим, що всю цитоплазму з мітохондріями, що містяться в ній, нащадки отримують разом з яйцеклітиною, у той час як у сперматозоїдах цитоплазма практично відсутня. З цієї причини жінка з мітохондріальним захворюванням передає його всім своїм дітям, а хворий чоловік – ні.
У нормальних умовах усі мітохондрії у клітині мають однакову копію ДНК - гомоплазмія. Однак у мітохондріальному геномі можуть відбуватися мутації, і внаслідок паралельного існування мутованої та немутованої мтДНК виникає гетероплазмія.
Наразі відомо більше 200 захворювань, викликаних мутацією мтДНК.
Клінічні прояви мітохондріальної патології:
У випадках, коли людина з мутацією в мітохондріальному гені несе суміш нормальної та мутантної ДНК - мутації спочатку можуть взагалі не мати зовнішніх проявів. Нормальні мітохондрії до певного часу забезпечують клітини енергією, компенсуючи недостатність функції мітохондрій з дефектами. Насправді це проявляється більш менш тривалим безсимптомним періодом при багатьох мітохондріальних захворюваннях. Проте рано чи пізно настає момент, коли дефектні форми накопичуються у кількості, достатньому прояви патологічних ознак. Вік маніфестації захворювання варіює у різних хворих. Ранній початок захворювання призводить до більш важкого перебігу та невтішного прогнозу.
Мітохондріальні мутації проявляються широким рядом клінічних симптомів. Ці мутації здатні залучати тРНК, рРНК або структурні гени і можуть бути біохімічно виражатися як дефекти всього електронно-транспортного ланцюга або як дефекти окремих ензимів. Мітохондріальні цитопатії вражають множинні органніістеми, але, як вказувалося, переважно уражаються органи з високою метаболічною активністю – мозок та скелетні м'язи. Таким чином, скелетні м'язи є тканиною вибору виявлення мітохондріальних хвороб.
Характерні ознаки мітохондріальних цитопатій:
- скелетні м'язи: низька толерантність до фізичного навантаження, гіпотонія, проксимальна міопатія, що включає фаціальні та фарингеальні м'язи, офтальмопарез, птоз;
- серце: порушення серцевого ритму, гіпертрофічна міокардіопатія;
- центральна нервова система: атрофія зорового нерва, пігментна ретинопатія, міоклонус, деменція, інсультоподібні епізоди, розлади психіки;
- периферична нервова система: аксональна нейропатія, порушення рухової функції гастроінтестинального тракту;
- ендокринна система: діабет, гіпопаратиреоїдизм, порушення екзокринної функції панкреасу, низьке зростання.
Гени, що входять до панелі:
| Ген |
Асоційований фенотип |
Спадкоємність.* |
ClinVar** |
HGMD** |
|
MT-ATP6
|
Нейропатія, атаксія та пігментний ретиніт, спадкова оптична невропатія Лебера, атаксія та полінейропатія, з початком у дорослому віці, кардіоміопатія, інфантильна гіпертрофічна, синдром Лі, стріатонігральна дегенерація, інфантильна, мітохондріальна
| Mitochondrial |
19 |
|
|
MT-ATP8
|
Кардіоміопатія, апікальна гіпертрофічна та нейропатія, Кардіоміопатія, інфантильна гіпертрофічна | Mitochondrial |
4 |
|
|
MT-CO1
|
Міоглобінурія, рецидивна, спадкова оптична нейропатія Лебера, сидеробластна анемія, дефіцит цитохром-С-оксидази, глухота, мітохондріальна |
Mitochondrial |
17 |
|
|
MT-CO2
|
Дефіцит цитохром C оксидази |
Mitochondrial |
8 |
|
|
MT-CO3
|
Дефіцит цитохром C оксидази, спадкова оптична нейропатія Лебера | Mitochondrial |
9 |
|
|
MT-CYB
|
|
Mitochondrial |
69 |
|
|
MT-ND1
|
Мітохондріальна міопатія, енцефалопатія, лактоацидоз та інсультоподібні епізоди, спадкова оптична нейропатія Лебера, атрофія та дистонія зорового нерва Лебера |
Mitochondrial |
21 |
|
|
MT-ND2
|
Спадкова оптична нейропатія Лебера, дефіцит мітохондріального комплексу I |
Mitochondrial |
6 |
|
|
MT-ND3
|
Атрофія та дистонія зорового нерва Лебера, дефіцит мітохондріального комплексу I |
Mitochondrial |
7 |
|
|
MT-ND4
|
Спадкова оптична нейропатія Лебера, атрофія та дистонія зорового нерва Лебера, дефіцит мітохондріального комплексу I |
Mitochondrial |
11 |
|
|
MT-ND4L
|
Спадкова оптична нейропатія Лебера |
Mitochondrial |
2 |
|
|
MT-ND5
|
Міоклонічна епілепсія з рваними червоними волокнами, мітохондріальна міопатія, енцефалопатія, лактоацидоз та інсультоподібні епізоди, спадкова оптична нейропатія Лебера, дефіцит мітохондріального комплексу I |
Mitochondrial |
19 |
|
|
MT-ND6
|
Мітохондріальна міопатія, енцефалопатія, лактоацидоз та інсультоподібні епізоди, онкоцитома, спадкова оптична нейропатія Лебера, атрофія та дистонія зорового нерва Лебера, дефіцит мітохондріального комплексу I |
Mitochondrial |
16 |
|
|
MT-RNR1
|
Глухота, мітохондріальна |
Mitochondrial |
3 |
|
|
MT-RNR2
|
Токсичність/резистентність до хлорамфеніколу |
Mitochondrial |
2 |
|
|
MT-TA
|
|
Mitochondrial |
4 |
|
|
MT-TC
|
Мітохондріальна міопатія, енцефалопатія, лактоацидоз та інсультоподібні епізоди | Mitochondrial |
3 |
|
|
MT-TD
|
|
Mitochondrial |
1 |
|
|
MT-TE
|
Діабет-синдром глухоти, Мітохондріальна міопатія, інфантильна, транзиторна, Мітохондріальна міопатія при цукровому діабеті |
Mitochondrial |
5 |
|
|
MT-TF
|
Міоклонічна епілепсія з рваними червоними волокнами, нефропатія, тубулоінтерстиціальна, енцефалопатія, мітохондріальна, епілепсія, мітохондріальна, міопатія, мітохондріальна, мітохондріальна енцефаломіопатія з ілактоацид
| Mitochondrial |
7 |
|
|
MT-TG
|
|
Mitochondrial |
3 |
|
|
MT-TH
|
|
Mitochondrial |
4 |
|
|
MT-TI
|
|
Mitochondrial |
7 |
|
|
MT-TK
|
Міоклонічна епілепсія з рваними червоними волокнами, синдром Лея |
Mitochondrial |
5 |
|
|
MT-TL1
|
Дефіцит цитохром-с-оксидази, міоклонічна епілепсія з рваними червоними волокнами, мітохондріальна міопатія, енцефалопатія, лактоацидоз та інсультоподібні епізоди, діабет-синдром глухоти, синдром циклічного блювання, СВДС
| Mitochondrial |
14 |
|
|
MT-TL2
|
Мітохондріальне мультисистемне захворювання, прогресуюча зовнішня офтальмоплегія, мітохондріальна міопатія, мітохондріальна енцефаломіопатія з лактоацидозом та інсультоподібними епізодами |
Mitochondrial |
5 |
|
|
MT-TM
|
Синдром Лея, мітохондріальний мультисистемний розлад |
Mitochondrial |
1 |
|
|
MT-TN
|
Прогресуюча зовнішня офтальмоплегія, мітохондріальне мультисистемне захворювання |
Mitochondrial |
3 |
|
|
MT-TP
|
|
Mitochondrial |
2 |
|
|
MT-TQ
|
Мітохондріальний мультисистемний розлад |
Mitochondrial |
2 |
|
|
MT-TR
|
Енцефалопатія, мітохондріальна |
Mitochondrial |
2 |
|
|
MT-TS1
|
Міоклонічна епілепсія з рваними червоними волокнами, мітохондріальна міопатія, енцефалопатія, лактоацидоз та інсультоподібні епізоди |
Mitochondrial |
10 |
|
|
MT-TS2
|
Мітохондріальний мультисистемний розлад |
Mitochondrial |
2 |
|
|
MT-TT
|
|
Mitochondrial |
5 |
|
|
MT-TV
|
Гіпертрофічна кардіоміопатія (ГКМП), синдром Лі, мітохондріальне мультисистемне захворювання, мітохондріальна енцефаломіопатія з лактоацидозом та інсультоподібними епізодами |
Mitochondrial |
3 |
|
|
MT-TW
|
Синдром Лея, міопатія, мітохондріальна |
Mitochondrial |
8 |
|
|
MT-TY
|
Мітохондріальний мультисистемний розлад |
Mitochondrial |
4 |
|
* Тип успадкування: аутосомно-домінантний (AD), аутосомно-рецесивний (AR), мітохондріальний (mi), Х-зчеплений (XL), Х-зчеплений домінантний (XLD) та Х-зчеплений рецесивний (XLR);
** ClinVar, HGMD - кількість варіантів гена, класифікованих як патогенні або ймовірно патогенні в базі даних ClinVar, HGMD.
Переваги цього тесту
- Лабораторія (США), акредитована САР
- Персонал, сертифікований CLIA, проводить клінічні випробування у лабораторії, сертифікованій CLIA.
- Потужні технології секвенування, передові методи збагачення мішеней та конвеєри точної біоінформатики забезпечують чудову аналітичну продуктивність.
- Ретельне складання клінічно ефективних та науково обґрунтованих генних панелей
- Загальнодоступна аналітична перевірка, яка демонструє повну інформацію щодо продуктивності тесту
- Аналіз некодуючих варіантів, що викликають захворювання
- Строга схема класифікації варіантів
- Систематичний робочий процес клінічної інтерпретації з використанням пропрієтарного програмного забезпечення, що забезпечує точну та простежувану обробку даних NGS.
- Докладний клінічний висновок
Як пройти дослідження
Вимоги здачі крові натще немає (легкий сніданок і незадовго до взяття крові випити 1-2 склянки звичайної негазованої води; дітей обов'язково напувати негазованою водою порціями, до 150-200 мл, протягом 30 хвилин), в день здачі крові не вживати медикаменти (включаючи вітаміни та БАД), напередодні виключити прийом смаженої та жирної їжі, виключити за 32 години алкоголь.
Проводиться забір венозної крові в пробірку з ЕДТА 4 мл.
Необхідно надати (взяти з собою або надіслати нам на електронну пошту) висновок лікаря з детальною клінічною картиною (опис симтомів, діагноз/попередній діагноз) та фото досліджуваного пацієнта (якщо є фенотипно значущі особливості), заповнити в електронному вигляді анкету.
У всіх випадках за результатами секвенування необхідно отримати консультацію лікаря-генетика.

 Київ
Київ
