Наследственная моторно-сенсорная невропатия (болезнь Шарко-Мари-Тута, ШМТ) тип Ia (CMT1A) — это самый распространённый демиелинизирующий подтип ШМТ, относящийся к наследственным моторно-сенсорным невропатиям. Характеризуется медленно прогрессирующей слабостью и атрофией дистальных мышц, нарушением чувствительности, снижением или отсутствием сухожильных рефлексов, а также деформациями стоп (высокий свод, молоткообразные пальцы). Начало чаще в детском или подростковом возрасте; продолжительность жизни, как правило, не сокращается.
Основная причина CMT1A — дупликация участка 17p11.2 (~1,4–1,5 Мб), содержащего ген PMP22. Избыточная экспрессия белка периферической миелиновой оболочки 22 (PMP22) нарушает формирование и поддержание миелина в периферических нервах, что приводит к демиелинизации и вторичной аксональной утрате. Тип наследования —
аутосомно-доминантный.
Клинические проявления
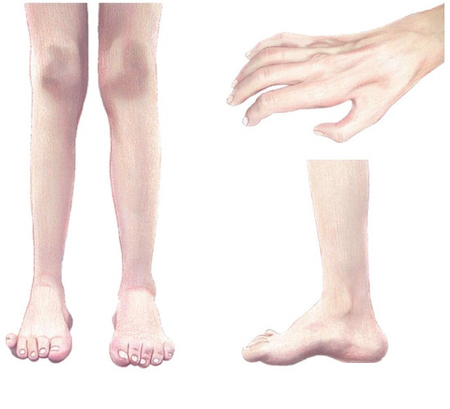
- Дистальная мышечная слабость и атрофия стоп и голеней, позже — кистей.
- Нарушение поверхностной чувствительности (онемение, парестезии), снижение вибрационной чувствительности.
- Гипо-/арефлексия ахилловых и коленных рефлексов.
- Деформации стоп: pes cavus (высокий свод), молоткообразные пальцы; частые «подворачивания» голеностопных суставов.
- Постепенное, обычно медленное течение с вариабельной выраженностью.
Диагностика
- Нейрофизиология: значительное снижение скорости проведения возбуждения по двигательным нервам (<38 м/с) для демиелинизирующих форм.
- Генетическое подтверждение: выявление дупликации PMP22.
- Клинический осмотр: оценка силы, рефлексов, походки, ортопедических деформаций; семейный анамнез.
Лечение и реабилитация
Каузальной терапии на данный момент нет; лечение поддерживающее:
- Физическая и трудовая терапия, упражнения на равновесие и растяжку.
- Ортезы, индивидуальные стельки, ортопедическая обувь; при показаниях — ортопедические вмешательства.
- Контроль боли и нейропатических симптомов; профилактика падений.
- Наблюдение мультидисциплинарной командой (невролог, реабилитолог, ортопед, подолог).
Прогноз
Течение медленно прогрессирующее; у большинства пациентов сохраняется
нормальная продолжительность жизни и возможность активной деятельности при условии
надлежащей реабилитации и ортопедической поддержки.
Ключевые факты
- Распространённость: CМТ в целом ~1:2 500; CMT1A — самый частый подтип CMT.
- Ген:
PMP22 (17p11.2), дупликация.
- Наследование: аутосомно-доминантное.
- Дебют: обычно детство/подростковый возраст.
Метод исследования
Анализ STR-полиморфизмов: D17S2226, D17S2227, D17S921 и D17S1358 хромосомной области 17р11.2-12 с целью выявления 1А-дупликации/ HNPP-делеции. В случае отсутствия информативности анализа (примерно 2–5% случаев), проводится прямое определение количества копий гена PMP22 с использованием ПЦР в реальном времени.
Другие анализы при Шарко-Мари-Тута
Для поиска генетической причины всех форм болезни Шарко-Мари-Тута рекомендуется Панель Шарко-Мари-Тута 153 гена.
 Киев
Киев
