Спадкова моторно-сенсорна нейропатія (хвороба Шарко-Марі-Тута, ШМТ) тип Ia (CMT1A) — це найпоширеніший демієлінізуючий підтип ШМТ, що належить до спадкових моторно-сенсорних нейропатій. Характеризується повільно прогресуючою слабкістю та атрофією дистальних м’язів, порушенням чутливості, зниженням або відсутністю сухожильних рефлексів, а також деформаціями стоп (високий звід, молоткоподібні пальці). Початок частіше у дитячому або підлітковому віці; тривалість життя зазвичай не скорочується.
Основна причина CMT1A — дуплікація ділянки 17p11.2 (~1.4–1.5 Мб), що містить ген PMP22. Надлишкова експресія білка периферичної мієлінової оболонки 22 (PMP22) порушує формування та підтримку мієліну в периферичних нервах, що веде до демієлінізації та вторинної аксональної втрати. Тип успадкування —
аутосомно-домінантний.
Клінічні прояви
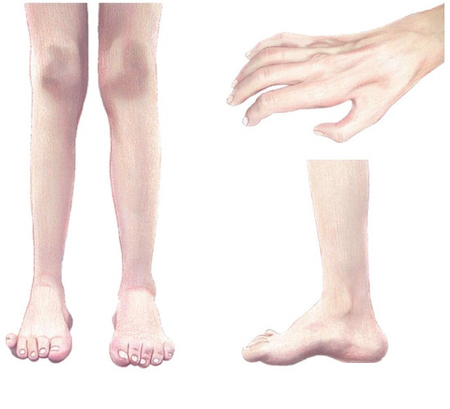
- Дистальна м’язова слабкість і атрофія стоп та гомілок, пізніше — кистей.
- Порушення поверхневої чутливості (оніміння, парестезії), зниження вібраційної чутливості.
- Гіпо-/арефлексія ахіллових і колінних рефлексів.
- Деформації стоп: pes cavus (високий звід), молоткоподібні пальці; часті «підвертання» гомілковостопних суглобів.
- Поступовий, зазвичай повільний перебіг з варіабельною вираженістю.
Діагностика
- Нейрофізіологія: значне зниження швидкості проведення збудження руховими нервами (<38 м/с) для демієлінізуючих форм.
- Генетичне підтвердження: виявлення дуплікації PMP22.
- Клінічний огляд: оцінка сили, рефлексів, ходи, ортопедичних деформацій; сімейний анамнез.
Лікування та реабілітація
Каузальної терапії наразі немає; лікування підтримувальне:
- Фізична та трудова терапія, вправи на рівновагу й розтягування.
- Ортези, індивідуальні устілки, ортопедичне взуття; за показаннями — ортопедичні втручання.
- Контроль болю та нейропатичних симптомів; профілактика падінь.
- Спостереження у мультидисциплінарній команді (невролог, реабілітолог, ортопед, подолог).
Прогноз
Перебіг повільно прогресуючий; у більшості пацієнтів зберігається
нормальна тривалість життя та можливість активної діяльності за умови
належної реабілітації та ортопедичної підтримки.
Ключові факти
- Поширеність: CМТ загалом ~1:2 500; CMT1A — найчастіший підтип CMT.
- Ген:
PMP22 (17p11.2), дуплікація.
- Успадкування: аутосомно-домінантне.
- Дебют: зазвичай дитинство/підлітковий вік.
Метод дослідження
Аналіз STR-поліморфізмів: D17S2226, D17S2227, D17S921 та D17S1358 хромосомної області 17р11.2-12 з метою виявлення 1А-дуплікації/ HNPP-делеції. У випадку відсутності інформативності аналізу (приблизно 2-5% випадків), проводиться пряме визначення кількості копій гена РМР22 із використанням ПЛР у реальному часі
Інші аналізи при Шарко-Марі-Тута
Для пошуку генетичної причини всіх форм хвороби Шарко-Марі-Тута рекомендується Панель Шарко-Марі-Тута 153 гени.

 Київ
Київ
