Панель з 71 гена, що включає оцінку варіантів, що не кодують.
Ідеально підходить для пацієнтів із клінічною підозрою на спадкове порушення згортання крові.
Не рекомендується пацієнтам з підозрою на тяжку форму гемофілії А, якщо інверсії не виключені з попередніх досліджень. Інверсія інтрону 22 гена F8 виявляється у 43–45% пацієнтів з тяжкою формою гемофілії А, а інверсія інтрону 1 – у 2–5% Цей тест не дозволяє надійно виявити ці інверсії.
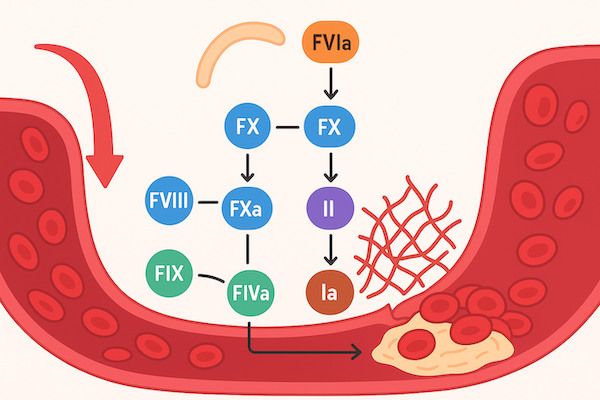
Розлади згортання крові – це гетерогенна група захворювань, спричинених дефіцитом функції тромбоцитів або факторів згортання крові. Розлади згортання крові можна розділити на три групи:
1) поширені спадкові розлади згортання крові, гемофілія A, B та хвороба фон Віллебранда (VWD);
2) рідкісні спадкові дефіцити факторів згортання крові;
3) спадкові розлади тромбоцитів.
VWD є найпоширенішим спадковим розладом згортання крові, що вражає до 1% загальної популяції та зустрічається з однаковою частотою серед чоловіків та жінок. Фенотипи, що охоплюються панеллю, включають VWD, гемофілію A та B, рідкісні розлади згортання крові, синдром Германського-Пудляка, синдром Віскотта-Олдріча, синдром Бернара Сульє, хворобу Гланцмана, тромбоцитопенію 2, сімейний тромбоцитарний синдром зі схильністю до гострого мієлолейкозу та синдром сірих тромбоцитів.
Молекулярні знання, отримані в результаті генетичного тестування, наразі регулярно використовуються в клінічній допомозі пацієнтам зі спадковим розладом згортання крові.
Гени, що входять до панелі:
| Ген |
Асоційований фенотип |
Спадкування* |
ClinVar** |
HGMD** |
|
ABCG5
|
Сітостеролемія |
AR |
13 |
42 |
|
ABCG8
|
Сітостеролемія |
AR |
18 |
44 |
|
ACTN1
|
Порушення згортання крові, тромбоцитарний тип 15 |
AD |
7 |
25 |
|
ADAMTS13
|
Синдром Шульмана-Апшоу, тромботична тромбоцитопенічна пурпура, сімейна |
AR |
30 |
183 |
|
ANKRD26
|
Тромбоцитопенія |
AD |
6 |
21 |
|
AP3B1
|
синдром Німецького-Пудлака |
AR |
14 |
34 |
|
ARPC1B
|
Аномалії тромбоцитів з еозинофілією та імуноопосередкованим запальним захворюванням |
AR |
2 |
4 |
|
BLOC1S3
|
синдром Німецького-Пудлака |
AR |
2 |
4 |
|
BLOC1S6
|
синдром Німецького-Пудлака |
AR |
1 |
2 |
|
CYCS
|
Тромбоцитопенія |
AD |
2 |
3 |
|
DTNBP1
|
синдром Німецького-Пудлака |
AR |
2 |
3 |
|
EFL1
|
Синдром Швахмана-Даймонда |
|
3 |
2 |
|
ETV6
|
Тромбоцитопенія 5 |
AD |
10 |
38 |
|
F10
|
Дефіцит фактора X |
AR |
15 |
155 |
|
F11
|
Дефіцит фактора XI |
AD/AR |
77 |
271 |
|
F12
|
Ангіоневротичний набряк, дефіцит фактора XII |
AD/AR |
7 |
53 |
|
F13A1
|
Дефіцит фактора XIIIA |
AR |
20 |
180 |
|
F2
|
Тромбофілія, спричинена дефектом тромбіну, дефіцитом протромбіну, вроджена |
AD/AR |
14 |
66 |
|
F5
|
Дефіцит фактора V, тромбофілія через резистентність до активованого протеїну C |
AD/AR |
19 |
157 |
|
F7
|
Дефіцит фактора VII |
AR |
27 |
322 |
|
F8
|
Гемофілія А |
XL |
296 |
3205 |
|
F9
|
Гемофілія B, чутливість до варфарину, тромбофілія, обумовлена дефектом фактора IX |
XL |
117 |
1281 |
|
FGA
|
Афібриногенемія, вроджена, Дисфібриногенемія, вроджена, Гіподисфібриногенемія, вроджена, Сімейний вісцеральний амілоїдоз |
AD/AR |
10 |
144 |
|
FGB
|
Афібриногенемія, вроджена, Дисфібриногенемія, вроджена, Гіподисфібриногенемія, вродженаенна |
AD/AR |
6 |
92 |
|
FGG
|
Афібриногенемія, вроджена, Гіподисфібриногенемія, Дисфібриногенемія, вроджена, Гіподисфібриногенемія, вроджена |
AD/AR |
7 |
127 |
|
FLI1
|
Тромбоцитопенія, тип Парі-Труссо, порушення згортання крові, тромбоцитарний тип 21 |
AD |
7 |
7 |
|
FLNA
|
Фронтометафізарна дисплазія, остеодисплазія Мельника-Нідлса, отопалатодигітальний синдром 1-го типу, отопалатодигітальний синдром 2-го типу, термінальна кісткова дисплазія з пігментними дефектами, перивентрикулярна вузликова гетеротопія 1-го типу, псевдообструкція, нейрональна, зчеплена з Х-хромосомою/вроджений синдром короткої кишки, дисплазія клапанів серця, зчеплена з Х-хромосомою |
XL |
133 |
257 |
|
FYB
|
Тромбоцитопенія 3 |
AR |
2 |
2 |
|
GATA1
|
Анемія без тромбоцитопенії, Тромбоцитопенія з бета-таласемією, Дизеритропоетична анемія з тромбоцитопенією |
XL |
21 |
15 |
|
GBA
|
хвороба Гоше |
AR |
84 |
488 |
|
GFI1B
|
Порушення згортання крові, тромбоцитарного типу, 17 |
AD |
6 |
9 |
|
GGCX
|
Розлад, подібний до псевдоксантоми еластичної, з множинним дефіцитом факторів згортання крові, факторів згортання крові, що залежать від вітаміну К, комбінований дефіцит |
AD/AR/Digenic |
13 |
42 |
|
GP1BA
|
Псевдохвороба Віллебранда, синдром Бернара-Сульє |
AD/AR |
9 |
73 |
|
GP1BB
|
Гігантське тромбоцитарне захворювання, ізольоване, синдром Бернара-Сульє |
AD/AR |
5 |
53 |
|
GP9
|
синдром Бернара-Сульє |
AR |
6 |
42 |
|
HOXA11
|
Радіоульнарний синостоз з амегакаріоцитарною тромбоцитопенією |
AD |
1 |
1 |
|
HPS1
|
синдром Німецького-Пудлака |
AR |
28 |
55 |
|
HPS3
|
синдром Німецького-Пудлака |
AR |
10 |
17 |
|
HPS4
|
синдром Німецького-Пудлака |
AR |
16 |
22 |
|
HPS5
|
синдром Німецького-Пудлака |
AR |
20 |
31 |
|
HPS6
|
синдром Німецького-Пудлака |
AR |
13 |
37 |
|
ITGA2
|
Фетальна та неонатальна алоімунна тромбоцитопенія |
AD/AR |
|
5 |
|
ITGA2B
|
тромбастенія Гланцмана |
AD/AR |
22 |
234 |
|
ITGB3
|
Порушення згортання крові, тромбоцитарний тип 15, тромбоцитопенія, неонатальна алоімунна, тромбастенія Гланцмана |
AD/AR |
18 |
165 |
|
LMAN1
|
Комбінований дефіцит факторів V та VIII |
AR |
5 |
37 |
|
MASTL
|
Тромбоцитопенія |
AD |
|
5 |
|
MCFD2
|
Фактор V та фактор VIII, комбінований дефіцит |
AR |
8 |
20 |
|
MECOM
|
Радіоульнарний синостоз з амегакаріоцитарною тромбоцитопенією 2 |
AD |
3 |
27 |
|
MPL
|
Тромбоцитемія, амегакаріоцитарна тромбоцитопенія |
AD/AR |
23 |
55 |
|
MYH9
|
Синдром Себастьяна, аномалія Мея-Хегглина, синдром Епштейна, синдром Фехтнера, макротромбоцитопенія та прогресуюча сенсоневральна глухота, глухота, аутосомно-домінантний тип успадкування 17 |
AD |
25 |
117 |
|
NBEAL2
|
Синдром сірих тромбоцитів |
AR |
10 |
51 |
|
P2RY12
|
Порушення згортання крові, тромбоцитарний тип 15 |
AD/AR |
4 |
13 |
|
PRKACG
|
Порушення згортання крові, тромбоцитарного типу, 19 |
AR |
1 |
1 |
|
PROC
|
Тромбофілія спадкова |
AD/AR |
36 |
387 |
|
PROS1
|
Тромбофілія спадкова |
AD/AR |
23 |
416 |
|
RASGRP2
|
Порушення згортання крові, тромбоцитарного типу, 18 |
AR |
3 |
20 |
|
RBM8A
|
Тромбоцитопенія - відсутність променевої кістки |
AR |
5 |
12 |
|
RUNX1
|
Сімейне порушення тромбоцитів із супутнім мієлоїдним злоякісним новоутворенням |
AD |
47 |
101 |
|
SERPINC1
|
Дефіцит антитромбіну III |
AD/AR |
44 |
412 |
|
SERPINF2
|
Дефіцит інгібітору альфа-2-плазміну |
AD/AR |
4 |
8 |
|
SLFN14
|
Тромбоцитопенія |
AD |
4 |
4 |
|
SRC
|
Тромбоцитопенія, аутосомно-домінантна, 6 |
AD |
2 |
1 |
|
SRP54
|
Синдром Швахмана-Даймонда |
AD |
3 |
|
|
TBXA2R
|
Порушення згортання крові, тромбоцитарний тип 15 |
AD |
1 |
6 |
|
THBD
|
Тромбофілія, спричинена дефектом тромбомодуліну, гемолітико-уремічний синдром, атипова |
AD |
5 |
28 |
|
THPO
|
Тромбоцитемія 1 |
AD/AR |
5 |
10 |
|
TUBB1
|
Макротромбоцитопенія |
AD | 2 |
7 |
|
VKORC1
|
Метаболізм лікарських засобів, пов'язаний з VKORC1, фактори зсідання крові, що залежать від вітаміну К, комбінований дефіцит |
AD/AR |
4 |
27 |
|
VWF
|
Хвороба Віллебранда |
AD/AR |
57 |
1009 |
|
WAS
|
Нейтропенія, тяжка вроджена, тромбоцитопенія, синдром Віскотта-Олдріча |
XL |
57 |
439 |
|
WIPF1
|
Синдром Віскотта-Олдріча 2 |
AR |
2 |
3 |
* Тип успадкування: аутосомно-домінантний (AD), аутосомно-рецесивний (AR), мітохондріальний (mi), Х-зчеплений (XL), Х-зчеплений домінантний (XLD) та Х-зчеплений рецесивний (XLR);
** ClinVar, HGMD - кількість варіантів гена, класифікованих як патогенні або ймовірно патогенні в базі даних ClinVar, HGMD.
Можлива персоналізація панелі
В панель дозволяється додати додаткові гени (до 200), пов'язані з симптомами, та видалити будь-які гени (щоб залишилося не менше 2). Вартість панелі може змінюватися, а саме:
- мала панель 2-25 генів ,
- середня панель 26-125 генів ,
- велика панель понад 126 генів .
Переваги цього тесту
- Лабораторія (США), акредитована САР
- Персонал, сертифікований CLIA, проводить клінічні випробування у лабораторії, сертифікованій CLIA.
- Потужні технології секвенування, передові методи збагачення мішеней та біоінформатичні пайплайни забезпечують точну аналітичну продуктивність.
- Ретельне складання клінічно ефективних та науково обґрунтованих генних панелей
- Аналіз некодуючих варіантів, що викликають захворювання
- Строга схема класифікації варіантів
- Систематичний робочий процес клінічної інтерпретації з використанням пропрієтарного програмного забезпечення, що забезпечує точну та простежувану обробку даних NGS.
- Докладний клінічний висновок
- Можна замовити сирі дані секвенування в форматі.vcf
Що далі?
Якщо аналіз виявив варіанти, що є можливою причиною хвороби, можна перевірити наявність цих варіантів у батьків. Аналіз окремих мутацій по Сенгеру.
Якщо аналіз не виявив варіантів, які є можливою причиною хвороби, можна розширити тестування до повного екзома Розширення панелі до повного екзома. Крім того, можна отримати сирі дані і робити повторну оцінку кожні кілька років. Можливо, згодом науці стануть відомі нові генетичні чинники захворювання.
Як пройти дослідження
Натще не потрібно, рясно пити воду.
Проводиться забір венозної крові в пробірку з ЕДТА 4 мл.
Забір матеріалу у нас у центрі або можна передати нам пробірку ЕДТА, взяту у будь-якій лабораторії. Про можливість забору у дітей уточнювати додатково.
У наших пунктах забору крім Києва забор матеріалу коштує .
Необхідно надати висновок лікаря з клінічною картиною (опис симтомів, діагноз), заповнити в електронному вигляді анкету.
Аналіз не проводиться для здорових людей без симптомів, у цьому випадку рекомендуються скринінгові аналізи на носійство.
За результатами секвенування необхідно отримати консультацію лікаря-генетика.

 Київ
Київ
